
to design drugs with graph neural networks 💊
notes on the problems with drug design and how gnns can help. 2142 words.

The problem of drug discovery can be illustrated with this (slightly oversimplified) example. Imagine that a certain protein plays a crucial role in a given disease. To attempt to find a cure for this disease, you can try to inhibit the function of this protein. One of the ways in which you could do this is to try and identify a molecule that can bind in this very well-defined binding pocket. It’s not as simple as it sounds. You have a bunch of different criteria that you have to actually optimize in order to find the best molecule. The first criteria is actually finding a molecule that binds to the known protein which would eliminate a lot of molecules from the start.
Then, you’d want to make sure that the molecule you’re taking to the next step is very selective for the protein that you are targeting. You don’t want it to be able to bind to any other protein - just the one that it’s intended to bind to.
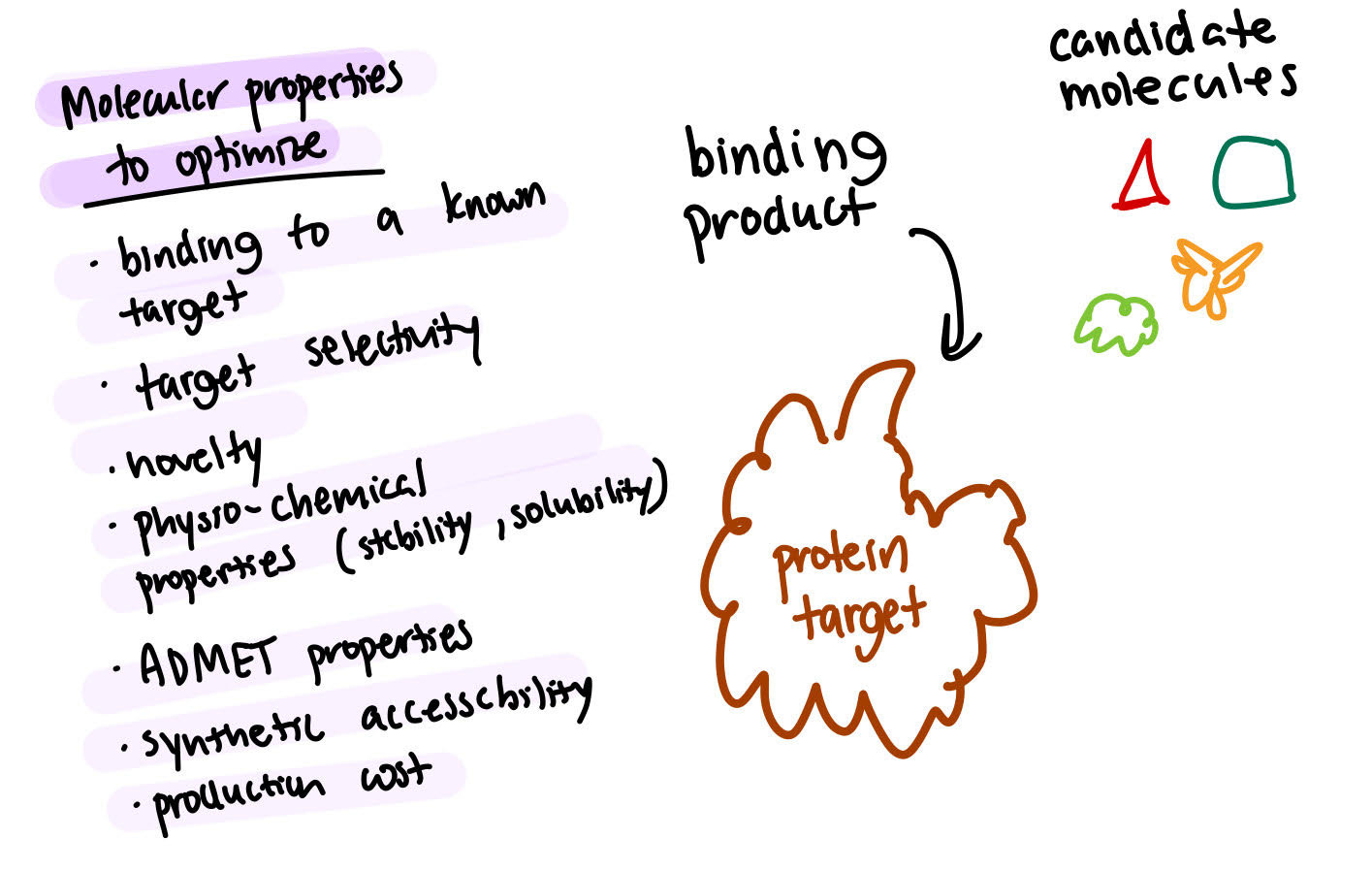
Maybe this means making some small modifications to the candidate molecule. The molecule should also be novel, so there may be a need for further modifications especially if it’s been previously patented. Optimizations and modifications can be made to improve the physical properties of the molecules. This is challenging because you have a very large number of interdependent properties that you’re trying to optimize over but also because there’s a large space of potential solutions. What makes this a difficult problem to solve is the fact that the space of theoretically possible drug-like molecules is between 10^20 to 10^60 number of molecules.
Traditionally, the drug discovery process involves screening large libraries of compounds. Now, scientists are proposing changes to these promising molecules through more novel methods of drug discovery. Communication to actually build novel drugs is a lengthy process as it involves many people as a result of the varying type of expertise needed. In fact, people spend a few years in the initial drug discovery phases. This is where scientists have become interested in how we can use deep learning to accelerate the drug discovery process.
Not only do we want to shorten the time that we spend in this initial drug discovery phase but we also would like to be able to find better solutions to increase the likelihood of success of the candidates that are tested in clinical trials.
molecular machine learning
Let's start with what a simplified molecular machine-learning pipeline looks like for molecular property prediction.
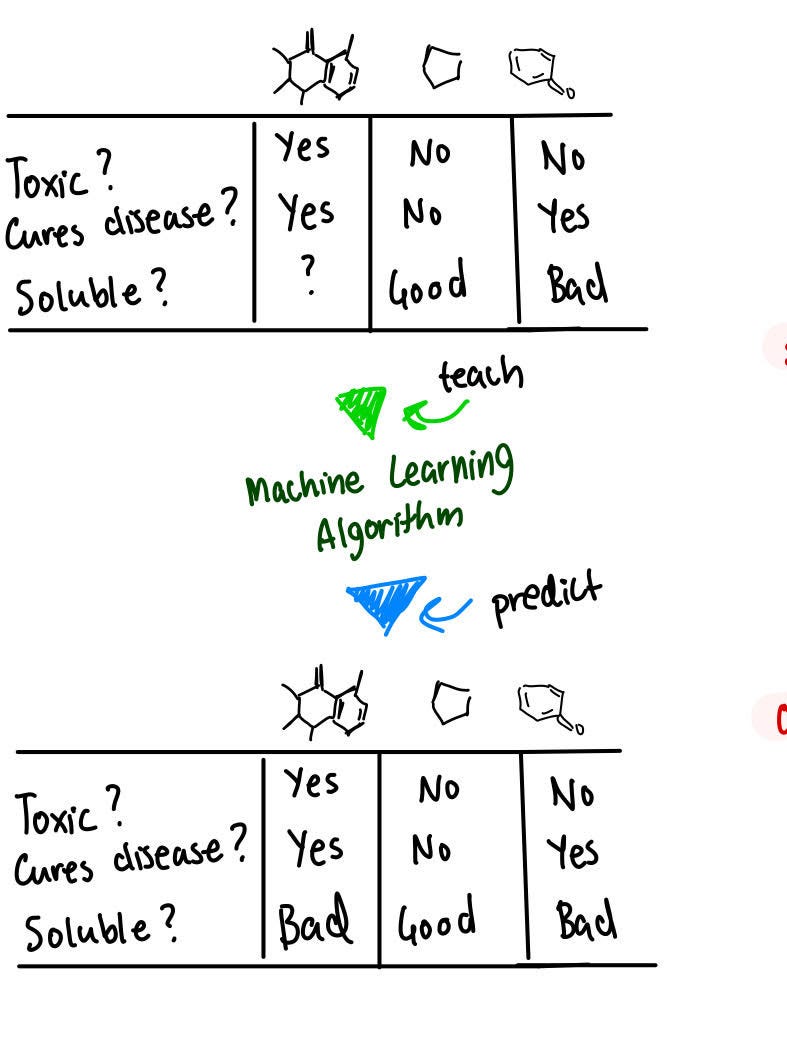
Having a set of different molecules and properties associated with each molecule means that you can take data and fit it into a machine-learning model. This model can help to predict the properties of other molecules that have not yet been seen.
Drug discovery becomes faster, more efficient, and more reliable when the focus is on property prediction. Property prediction, which is key to many other parts of drug discovery like screening and featurization can be done today, using graph neural networks.
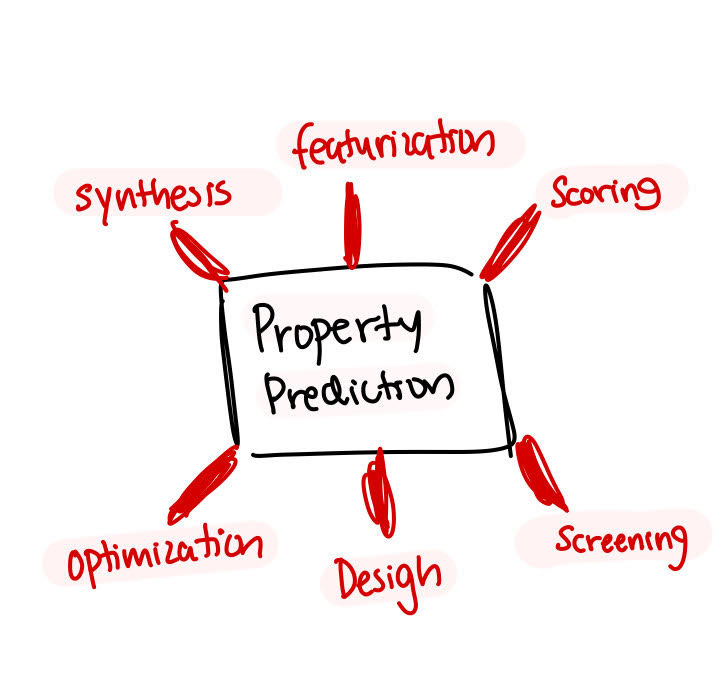
To understand why graph neural networks are ideal for drug discovery, we need to understand what a molecule is.
The definition of a molecule may change based on who you’re asking.
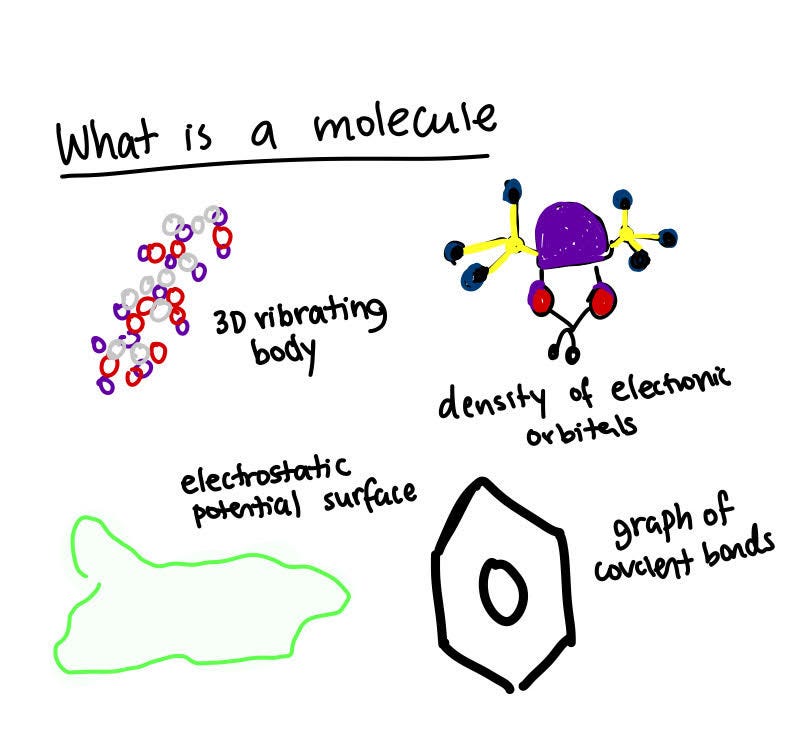
Some people would say that it's a 3D vibrating body that vibrates in space depending on the temperature and environment conditions.
Other people may say that it’s a density of electronic orbitals. The reactivity, shape, and structure of the molecule will depend on where the electrons are at a given time.
To decide the structure of the molecule, other people would say to look at the surface of the molecule and the electrostatic potential as it dictates how the molecule will interact.
learning a model
The question is, how do we learn a model to encompass all this knowledge to design molecules?
With geometric deep learning, we learn on graphs - the 2D structures on moving bodies and on surfaces. Geometric deep learning is very recent in deep learning, but it’s becoming more popular, especially in recent years.
Along with geometric deep learning, transfer learning can be used to learn on one task or representation and then transfer onto another one. You can easily imagine learning how this graph correlates with this 3D vibrating body, so there is essentially shared knowledge between them even though the representation is very different.
We can also imagine a form of meta-learning (I wrote an article on this earlier). We learn common knowledge across all tasks and representations. Ideally, we would like to be able to do zero-shot or few-shot predictions where we infer some properties without having data.
We just need to readjust the model using only a few data points and if we really understand what the molecule is, zero-shot and few-shot learning prediction becomes possible. For this, though, we need to have a deeper understanding of the molecule. One of the things that have been done in chemistry and drug discovery for a long time was the use of molecular fingerprints.
in summary 👇

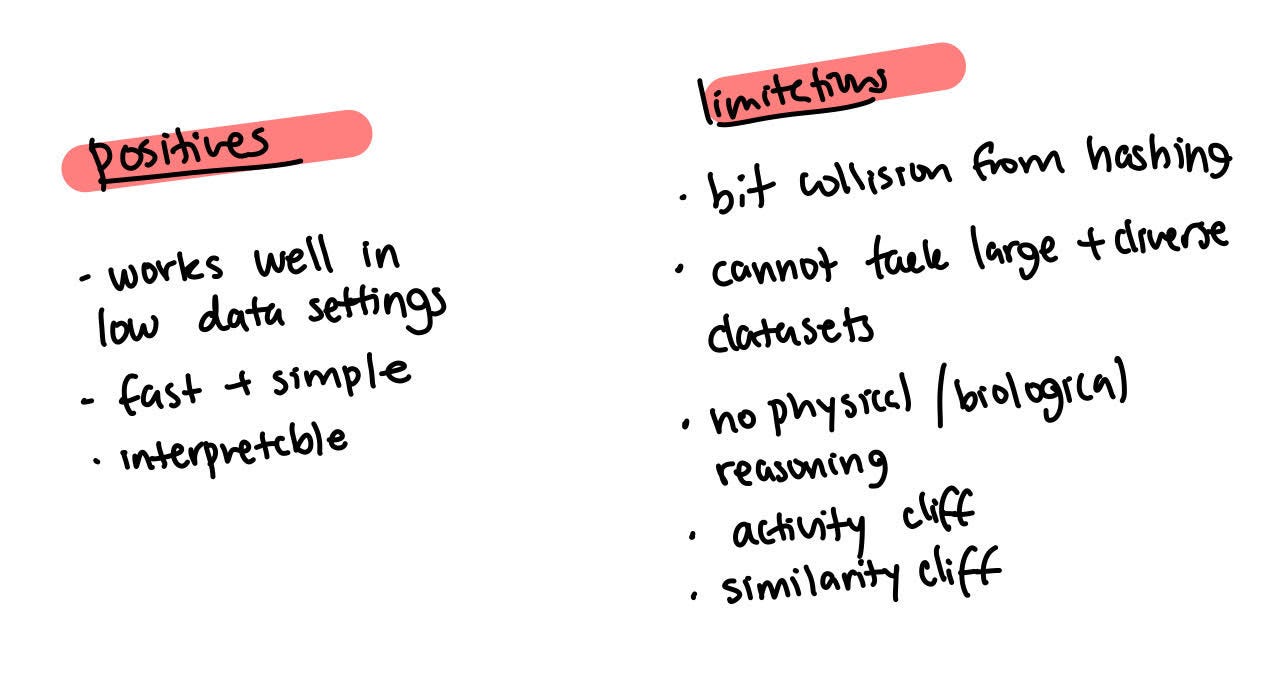
Imagine you take this molecule and you find all the fragments in the given molecule. You assign it a different bits but only three fragments are represented. In reality, there are other fragments at the intersection of each of these fragments. Surprisingly, though, this method has been very successful as it works well with settings in which there is no data. It’s fast, simple, interpretable, and also the first instance of graph machine learning.
This kind of fingerprint shortcut has some problems though. For example, it leads to a limited global understanding of the molecule because you have these piece fragments but you don’t really have ideas on how they’re placed together.
There’s also collision due to hashing which means it isn’t able to tackle large and diverse data sets. The bigger the data set, the bigger the fingerprint you need to have because each byte in the representation can be representative of different structures. Machine learning can’t tackle this problem. There isn’t any physical or biological reasoning. For example, a large part of the behaviour of a molecule may depend on which substructure is present.
Defining this can lead to some activity cliffs - tiny changes in the molecule that can lead to a large change in how the molecule behaves. This is because there’s no physical or biological understanding of each of these fragments that are just there. If we have electronic orbitals then we can see that small changes can have a larger change on these orbitals. Depending on where the change happens, it can affect many elements or even a single element of the vector.
Because of this limitation, the next step is to use graph machine learning where using the graph structure, we can not only learn a representation that is about which fragments are present, but we can also learn representations that contain knowledge about physics to biology. Moreover, we can compress the compressor substructure more intelligently in a way that is more learned.
the molecular maze
Across hundreds of videos and articles, the most commonly used analogy for drug discovery with deep learning is with mazes and graphs because they’re able to best explain the molecular graph.
When we want to do property prediction with graph neural networks, first we need to understand why GNNs are optimal. It’s arguably the simplest and most common and interpretable representation of a molecule. They don’t require simulation or experiments to be used so if you only have a single data set you can use them directly without requiring physical simulation or docking software.

In the simulation above, brute force methods of simulating drugs in new environments (through reinforcement learning) adapted to different bodily conditions can help test drug effects.
Working with graphs is similar to navigating a maze where each intersection of the maze can be seen as edges in this molecular graph. Many methods have focused on doing something that looks like a random walk. However, if you’re stuck in a maze and you simply walk randomly you’re going to stay stuck in the maze for a really long time. You need to really understand the structure of the maze to be able to navigate it as efficiently as possible. Of course, there are many strategies to navigating this maze:

You can walk and look around, but try not to just look around quickly to gather as much information as possible. This strategy can be used but it has problems. For example, it can lead to over-smooothing or a lack of expressivity.
You can also colour your way there. As soon as you find some structure then just colour that substructure and you’ll be able to navigate the maze more efficiently. If you have too many colours though (too many substructures), it can also become confusing.
You can squeeze through narrow paths. This is when the graph is very large. You need the information to pass from very distant neighbours. You have some very strong bottlenecks at some parts of the graph. They don’t really apply to standard small molecular graphs.
You can destroy the walls which will allow you to connect nodes that seem far away. The problem with this is you could destroy too many walls and the maze would fall apart.
Using a map, you can define positional encoding to navigate the graph much more efficiently without getting lost. You do have to make sure that the map that you have is a good representation and gives accurate information about your position. If your map is broken or if your map is wrong, you can get lost even more easily.
Of course, you don’t even need to navigate the maze if you have wings. In practice, this means a fully-connected graph transformer. The main downside to this strategy is that you need to learn the map before flying and you need lots of ‘energy’ to fly.
You could also ask yourself: why don’t we just connect parts in the graph that are very far apart finally listen to the shape of the maze so if you make some noise or knock on the walls, you can hear the resonance of the maze which can help you understand the structure better.
All the strategies are essentially just analogies of the different methods that have been developed to build graph neural networks and improve our understanding of drug discovery.
The maze represents molecular graphs.
#1, 5, and 6 are generally the most commonly used. Skipping over some theory and implementation to better explore in later posts but I think it’s valuable to know what this looks like in practice.
implications of molecular mazes
Let’s imagine ligand and receptor molecules that are represented as graphs containing the appropriate information. We need to pass this information into a graph neural network. The way it works is it passes the node and edge features through a shared hidden layer with softmax activation to build eighteen fingerprints which are sound enough to form a molecular fingerprint
Previous atom feature vectors are calculated from each atom feature vectors and information about its neighbours. The graph neural network collects the information of neighbouring nodes and edges to each node and edge recursively until the given depth is hit. Later the item and edge fingerprints of a particular depth is collected and passed through a dense layer.
It’s usually fit to some property. Imagine you have a graph and you want to have it as some vectors of some particular shape called embedded space.
Now, you have to create something - a mapping function from a graph of arbitrary size and shape to some predefined shape of embedding space you want to compute. In the diagram below you can see the neighbouring nodes being tracked in the bottom graph.

We cursively collect features from these nodes in a way that we multiply all the features of an appropriate node to some activation function.
If there are multiple nodes, we’re basically transforming into embedded space. We can do this by multiplying the value of features of this node for some weight mattress and then summing it up and passing it through an activation function like in a regular neural network. This information can be passed in the next step of recursion - we do that recursively until the given depth is attained.
There are generally two kinds of graph neural networks: convolutional and recurrent. The main difference is that in recurrent neural networks, we can’t apply the same set of weights until a convergence criterion is met. In convolutional neural networks, we apply different weights at each iteration.

Combining all the pieces of this picture, imagine you have a drug that is represented with the drug's chemical structure, smile.
The representation for SMILE can be changed into a graph and of course, the same can be done for the target protein as well.
You can extract a contact map from that. The contact map is a simple representation of a secondary structure of this target protein. Then you transform this contact map into a protein graph. Now, you have two graphs: one for a ligand and one for a receptor. You pass these graphs into graph neural networks. Later, you concatenate these two neural networks and fit them into affinity to create an accurate network that can find the right molecules.
I’m learning by writing about the projects I’m doing to improve my technical understanding. If you find errors in my understanding or would just like to talk, reach out @_anyasingh. You can also connect with me through links on my website.